Case Report
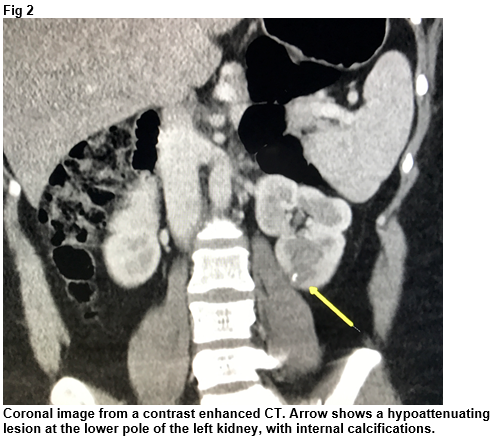
A 37-year-old female was referred by her general practitioner for a CT thorax, abdomen and pelvis to investigate long standing tiredness and upper GI symptoms. Of note, typical features of carcinoid syndrome were not present. She had a background history of euthyroid multinodular goitre, hiatus hernia, elevated BMI and anxiety. CT revealed a 1.4cm hypoattenuating lesion with internal calcifications at the lower pole of the left kidney (Fig 2). There was no evidence of metastatic disease on CT.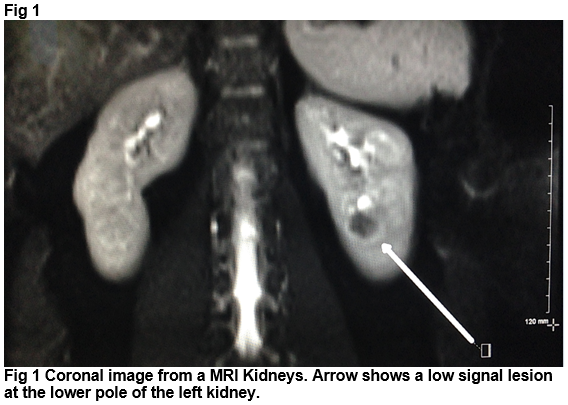
She was referred to urology and her case was discussed at a multidisciplinary team meeting at our institution. Given that the appearance of the lesion was atypical for conventional renal cell cancer, a MRI kidneys was performed (Fig 1). Following MDM discussion, a CT guided biopsy of this lesion was performed. The decision to proceed with biopsy was based on the atypical imaging features and the patient’s young age. Histology from the CT guided biopsy showed a solid and trabecular tumour composed of epithelioid cells with uniform round nuclei. These cells were positive for CD56 and chromogranin. There were no mitoses and the proliferation rate (as measured by MIB1 stain) was <two percent. The features were consistent with a well differentiated neuroendocrine tumour. She went on to have a laparascopic left nephrectomy. Histology of the surgical sample confirmed a well-differentiated neuroendocrine tumour. Ongoing clinical and radiological follow-up is planned, with repeat CT TAP booked for 12 months following surgery.
Discussion
Neuroendocrine tumours arise from cells within the endocrine and nervous systems, and comprise a heterogenous group of neoplasms with varying behaviours. They most commonly arise in the gastrointestinal tract, pancreas and lungs, and are generally distinguished based on anatomical location, well-differentiated (low and intermediate grade) and poorly differentiated (high grade) features. Carcinoid syndrome is a constellation of symptoms including flushing, diarrhoea, bronchoconstriction and occasional secondary restrictive cardiomyopathy. It results from the excessive production of serotonin and other vasoactive substances by the tumours, and usually occurs secondary to metastatic tumours which arise in the distal small bowel and proximal colon1. 24-hour urinary excretion of 5-HIAA, a metabolite of serotonin, can be measured to assess for carcinoid syndrome.
Diagnostic imaging of carcinoid tumours usually involves contrast enhanced CT or MRI. Lamb et al’s radiologic review reports CT and MRI features alone cannot reliably differentiate between renal NET and renal cell cancer, with no feature specific to renal carcinoid. Calcifications were common, however, seen in one third of their cases2. Octreotide scanning, which involves injection of Indium-111 pentetreotide, a radiolabelled somatostatin analogue, can be useful for initial staging and for follow-up imaging of some carcinoid tumours. Primary neuroendocrine tumours of the kidney are exceedingly rare, with less than 100 case reports published in the literature in 20163, 4. Many studies report a higher incidence in horseshoe kidneys5, with one retrospective study reporting that 22.3% of the 85 cases were in horseshoe kidneys2. Carcinoid syndrome is reported rarely with primary renal NETs, one study reporting an incidence of 4.7%2. Nephrectomy is currently the mainstay of treatment for renal carcinoid tumours. Prognosis varies depending on differentiation of the tumour and presence of metastases.
The imaging findings of an atypical renal lesion prompted us to consider pre-operative biopsy in this case. The presence of an atypical renal mass on imaging should always raise the suspicion for an alternative to renal cell cancer. Other lesions which can occur in the kidney, including oncocytoma and lymphoma, may not require nephrectomy. In such cases, and notably in young patients, biopsy prior to surgery should be strongly considered.
In summary, the presentation of atypical imaging findings of a renal mass should prompt consideration to biopsy. In the case we have presented, the biopsy confirmed the presence of a very rare primary renal carcinoid.
Conflicts Of Interest
There are no conflicts of interest regarding this paper
Correspondence:
Dr Maeve O'Sullivan, Radiology Department, AMNCH, Tallaght, Dublin 24
Email: [email protected]
References
1. Strosberg JR. Clinical features of the carcinoid syndrome. UpToDate [Internet]. 2015 Jul 21. Available from: https://www-uptodate-com.proxy.library.rcsi.ie/contents/clinical-features-of-the-carcinoid-syndrome?source=search_result&search=clinical%20features%20of%20the%20carcinoid%20syndrome&selectedTitle=1~74
2. Lamb L, Shaban W. Primary renal carcinoid tumor: A radiologic review. Radiol Case Rep. 2015 Dec 7;9(2):923. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27186242
3. Teegavarapu PS, Rao P, Matrana M, Cauley DH, Wood CG, Tannir NM. Neuroendocrine tumors of the kidney: a single institution experience. Clin Genitourin Cancer. 2014 Dec;12(6):422-7. Available from: https://www.ncbi.nlm.nih.gov/pubmed/?term=neuroendocrine+tumors+of+the+kidney%3A+a+single+institution+experience
4. Li B, Cui T, Ban Z, Luo L, Sun L. Primary renal carcinoid tumor: case report and review of the literature.Onco Targets Ther. 2016 Feb 23;9:741-3. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26966374
5. Wann C, John NT, Kumar RM. Primary renal large cell neuroendocrine carcinoma in a young man. J Clin Diagn Res. 2014 Nov;8(11):ND08-9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/?term=primary+renal+large+cell+neuroendocrine+carcinoma+in+a+young+man
P677