Introduction
Proliferative Myositis (PM) is a rare, benign tumour easily mistaken for sarcoma at presentation. Here, we propose that LD PM developed following repetitive rowing-induced myotrauma in a young male.
Case Report
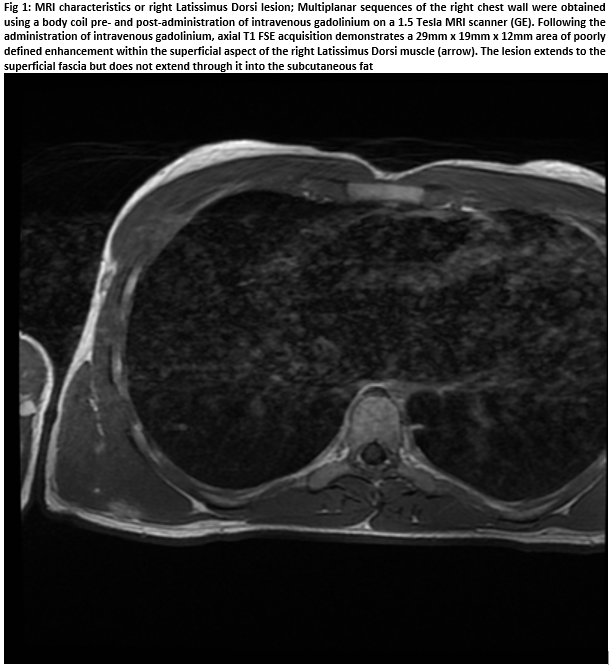
A 20-year-old male presented with an unresolving, tender subcutaneous mass on his chest wall of sudden development five months prior. Overlying skin unchanged. No history of trauma. However, the patient described rowing competitively in the previous months. No systemic disturbances. Clinical examination revealed a firm, discrete, deeply-seated solitary 2cm mass attached to underlying LD but untethered to overlying skin. Ultrasound demonstrated an intramuscular mass, warranting further imaging. MRI subsequently visualised a poorly-defined 29x19x12mm mass within the LD, abutting superficial fascia but with no subcutaneous fat extension (Figure 1).T2-weighted sequencing hyper-intensified the lesion, indicating oedematous tissue. An urgent trucut biopsy was obtained to diagnose/out-rule suspected sarcoma.
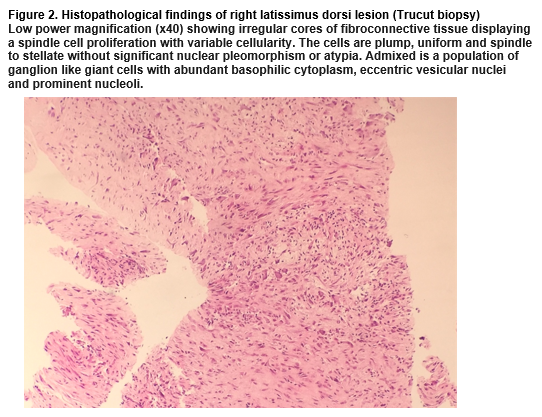
Histopathological examination revealed irregular cores of fibroconnective tissue displaying a spindle cell proliferation. The cells were plump, uniform and spindle to stellate without significant nuclear pleomorphism/atypia. Admixed was a population of ganglion-like giant cells with abundant basophilic cytoplasm, eccentric vesicular nuclei and prominent nucleoli (Figure 2). High power magnification demonstrated the co-existence of spindle and stellate fibroblasts and ganglion-like myofibroblasts. Spindle cells were CD68(+) and smooth muscle antigen(+) (supporting a fibroblastic process) and negative for desmin, beta-catenin and s100. Therefore, sarcoma or desmoid-type fibromatosis was unlikely. The constellation of findings was consistent with PM.
Discussion
PM is a rare, benign tumour easily confused with sarcoma at presentation due to its rapid growth within the skeletal muscle of predominantly middle-aged patients1,2. The median age of onset is 50 years with males slightly more affected than females1. Areas most commonly affected include the head, neck and upper extremities1,3. The rapid growth is commonly painful, however, lymphadenopathy and inflammation are usually absent3,4.
Although the aetiology of PM remains unknown, several theories have been proposed including muscular trauma2-5. Enzinger reported two cases discovered in mailmen who had complained of mailbag-induced muscle irritation preceding the development of the lesions1. As mentioned, our patient was a competitive rower who sustained repeated strain on his LD, supporting this theory. Alternative theories include local ischemia and paracrine myopathy4.
Following thorough history and examination, radiographic investigations are warranted. Ultrasonography is a valuable initial approach4. Pathognomonic findings on ultrasound include what has been described as a "Scaffolding" pattern in which hypoechoic geometric lines within dense, hyperechoic muscle are visualised4. Additional imaging modalities include CT and MRI. A poorly demarcated intramuscular lesson with irregular borders is typically demonstrated by CT. MRI appearances vary according to sequence. Hypointense/isointense lesions are generally demonstrated by T1-weighted sequencing whereas T2-weighted sequences may reveal strongly hyper-intensification. This is suggestive of inflammation5, as was the case with our patient.
Definitive diagnosis is made histopathologically. Tissue can be obtained by fine needle aspiration or incisional biopsy, aiming to visualise the characteristic "checkerboard" pattern of myofibroblasts infiltrating surrounding healthy muscle, ganglion-like basophilic giant cells and a lack of atypical mitosis3,4,5. Macroscopically, the lesions are frequently poorly-demarcated, appearing as an area of white scar-like induration of muscle.1 Diagnostically, PM falls within a spectrum of benign myofibroblastic tumour-like lesions, including modular fasciitis and proliferative fasciitis6.
Symptoms and rate of progression determine management of PM. Surveillance is the preferred option with reports suggesting PM tends to spontaneously regress, often with complete resolution achieved within months. If the tumour progresses, causes functional impairment or becomes cosmetically distressing, conservative surgical excision is recommended2-5. There is currently no role for radical resection as there have been no documented cases of recurrence or malignant transformation2-5.
Conflict of Interest:
The authors declare that there is no conflict of interests.
Correspondence:
Nathaniel Mc Hugh, Department of Plastic and Reconstructive Surgery, Cork University Hospital, Wilton, Cork, Ireland
References
1. Enzinger, F.M. & Dulcey, F. Proliferative myositis. Report of thirty-three cases. Cancer 20, 2213-2223 (1967).
2. Kunze, B., von Weyhern, C. & Kluba, T. Myositis proliferans: diagnosis and therapy of a pseudosarcomatous soft tissue lesion. Musculoskeletal surgery 95, 255-257 (2011).
3. Fauser, C., Nahrig, J., Niedermeyer, H.P. & Arnold, W. Proliferative myositis: a rare pseudomalignant tumor of the head and neck. Archives of otolaryngology--head & neck surgery 134, 437-440 (2008).
4. Colombo, J.R., Dagher, W. & Wein, R.O. Benign proliferative myositis of the sternohyoid muscle: review and case report. American journal of otolaryngology 36, 87-89 (2015).
5. Pagonidis, K., Raissaki, M. & Gourtsoyiannis, N. Proliferative myositis: value of imaging. Journal of computer assisted tomography 29, 108-111 (2005).
6. Kato, K., Ehara, S., Nishida, J. & Satoh, T. Rapid involution of proliferative fasciitis. Skeletal radiology 33, 300-302 (2004).
(P605)