Introduction
To date, numerous colorectal cancer and polyposis syndromes have been described, including familial adenomatous polyposis (FAP), Peutz-Jeghers syndrome, Juvenile polyposis syndrome and hereditary mixed polyposis. Polyposis syndromes confer a risk of malignancy as well as polyps, given that, irrespective of the genetic aetiology, changes in any individual polyp along the adenoma-carcinoma pathway may lead to the development of colorectal cancer, a risk that increases with increasing polyp burden. Lynch syndrome (Hereditary Non-Polyposis Colorectal Cancer syndrome) predisposes to colorectal and other cancers in the absence of preceding polyposis1.
MUTYH-associated polyposis (MAP) was the first recessively-inherited polyposis syndrome to be described2. The phenotype of MAP is similar to that of attenuated familial adenomatous polyposis (FAP), with colonic polyp burden ranging from tens to a few hundred, developing at a mean age of fifty. MAP is also associated with other gastrointestinal polyps, duodenal cancer3 and extra-intestinal malignancies, including ovary and bladder. There is also some conflicting evidence of an association with breast and endometrial cancers 3-5.
To date, 306 unique DNA variants in MUTYH have been detected in 8514 individuals worldwide6. In Northern European populations however, bi-allelic combinations of two common missense variants, c.536A>G (p.Tyr179Cys) in exon 7 and c.1187G>A(p.Gly396Asp) in exon 13, are reported in 70% of patients with MAP7, and at least one of these two mutations are detected in 90% cases. The combined carrier frequency of the two common mutations in unaffected controls of European origin ranges from 1 in 80 to 1 in 5007. Large ethnic variations in carrier frequency exist, however, and the two common missense mutations have not been reported with the same frequency in Asian 8, 9 or Jewish populations 10, suggesting a founder effect 7.
The majority of germline testing of MUTYH for patients in the Republic of Ireland is co-ordinated through a single molecular laboratory based in the Department of Clinical Genetics at Our Lady’s Children’s Hospital Crumlin. The aims of this study were to examine the incidence and impact of MYH mutations in Irish patients referred for genetic counselling/testing, to identify a genotype-phenotype association, and to investigate the clinical outcome of mono-allelic MUTYH mutation carriers.
Methods:
A retrospective cohort study was undertaken. The cohort under investigation included mono-allelic and bi-allelic MUTYH carriers identified on testing performed via the molecular laboratory in Our Lady’s Children’s Hospital Crumlin. Probands included Irish patients tested in Ireland; patients of non-Irish origin tested in Ireland, and Irish patients tested abroad where family members had attended for cascade testing. Patients referred for MUTYH testing between 2003 and February 2016 were identified by searching departmental electronic patient databases (iGene and Crumbase) using keywords “MUTYH” and “MYH”. Mutation testing was performed by real-time assay for two common European mutations, with full sequencing of the gene in the case of patients with convincing MAP phenotype with only mono-allelic or no mutation detected by rtPCR. Six probands with severe phenotype also underwent sequencing of the APC gene. Details regarding phenotype and genotype were obtained by chart review.
Results:
MUTYH analysis was performed in 120 patients from 61 families, including diagnostic testing in 58 probands. Seventeen unrelated probands were found to carry bi-allelic MUTYH mutations, including one patient of Portuguese ethnicity. One proband with a convincing phenotype underwent diagnostic testing for the two common European mutations only. One mutation has been identified, and full MUTYH sequencing is underway to identify the second mutation. During cascade screening, a further nine bi-allelic mutation carriers and twenty-nine mono-allelic mutation carriers were identified (Table 1).
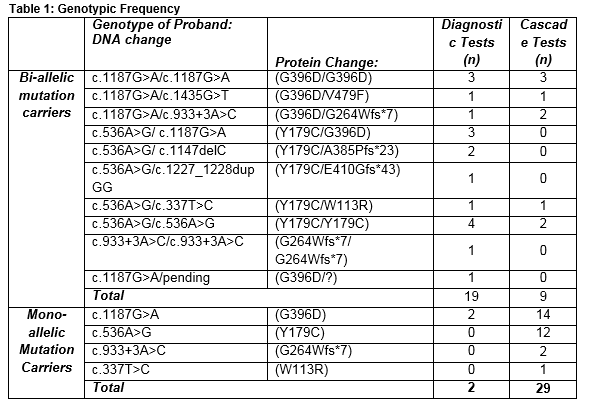
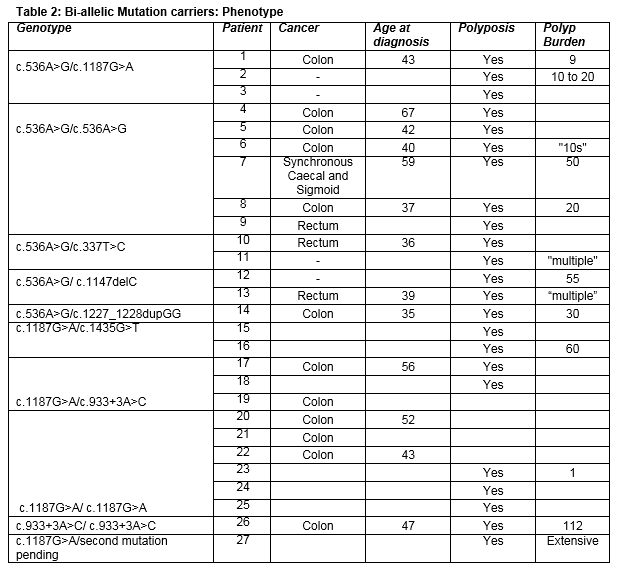
The mutations most commonly reported in our cohort were the two common European mutations, with c.536A>G homozygosity reported in four families; c.1187G>A homozygosity in three families; and compound heterozygosity of the two mutations in three families. Five families had compound heterozygote genotypes including at least one of the two common European mutations. All six bi-allelic c.536A>G homozygous mutation carriers were affected by cancer on a background of polyposis, at mean age 49 years (±13.2). Of six c.1187G>A homozygous mutation carriers, three siblings developed colon cancer at mean age 47.5 years (±6.4). The phenotypes of all bi-allelic mutation carriers are outlined in table 2. There were no cases of duodenal, ovarian, bladder, breast or endometrial cancers among bi- allelic mutation carriers in this series.
Two patients were found to carry variants of unknown significance after undergoing full MUTYH sequencing. One of these patients had ~12 polyps at age 49, and was found to carry two variants of uncertain significance, c.-2C>G (p.Ala2Pro) and c.1256C>A (p.Ala419Asp). Another patient presented with polyposis at age 79, and was found to carry a single MUTYH variant c.312C>T (p.Tyr104=).
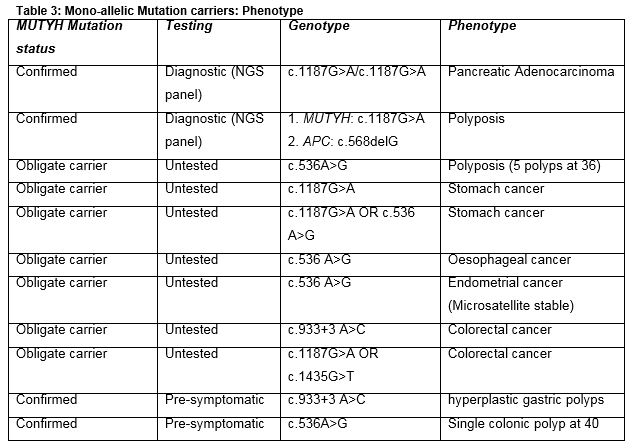
Diagnostic testing of patients for cancer predisposition using next-generation panels including MUTYH identified a number of mono-allelic mutation carriers. One patient underwent such testing following diagnosis of adenocarcinoma of the pancreas on a background of strong familial history of colorectal, ovarian, liver and pancreatic cancer. In this family, the MUTYH mutation was not shown to segregate with disease. Another index patient underwent panel testing for polyposis predisposition, and was found to carry a single paternally inherited MUTYH mutation (c.1187G>A) as well as a pathogenic de novo mutation in the APC gene (c.568delG, p.Glu190Asnfs*15). This patient had a classical phenotype of Familial Adenomatous Polyposis, with 250 adenomatous polyps on colonoscopy aged 21 years. There was no family history of polyposis or cancer in this instance. Several obligate carriers of MUTYH mutations were noted to have a previous history of malignancy, including of the stomach, oesophagus and endometrium as well as colorectal disease (Table 3). However the majority of obligate carriers did not report a history of malignancy or polyposis. Among mono-allelic MUTYH mutation carriers picked up on cascade testing (n=29), the majority had not undergone colonoscopy. Of those that did (n=8), seven had normal tests, and one had a solitary colonic polyp.
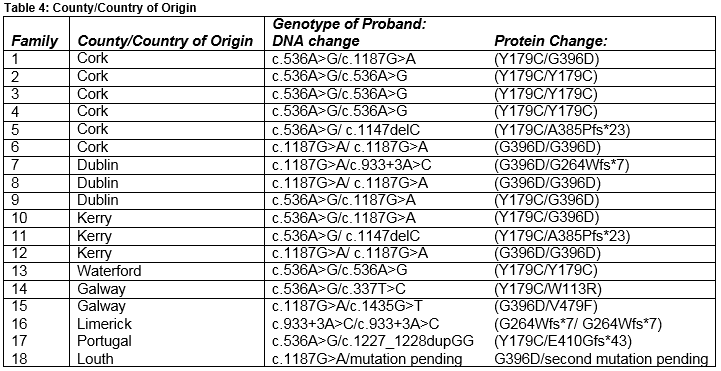
A local geographical difference in incidence of MUTYH-associated polyposis was noted, with marked predominance in the South of the country (table 4). Six of sixteen apparently unrelated families originated from Co. Cork, a further three from neighbouring Co. Kerry, and one from Co. Waterford. Another family originated in Co. Limerick, and two from Co. Galway. A disproportionately small number of families (n=3) originated from Co. Dublin, given the distribution of the population in Ireland. One family was of Portuguese origin and now live in Dublin. The rates of colorectal cancer nationally have also shown a regional variation, with observed incidence of colorectal cancers. Invasive cancers in Co. Cork have been higher than that expected in both periods between 1994-2003 and 2004-2012 11.
Discussion
Cancer arises as a result of sequential acquisition of genetic mutations, caused by exogenous or endogenous mutagens. There are intrinsic DNA repair mechanisms to repair such mutations, and MYH polyposis is a genetic disorder of one such DNA repair mechanism. DNA repair mechanisms have generated particular interest of late, given that last year’s Nobel Prize in Chemistry was jointly awarded to three renowned scientists in this field12.
In our series, c.536A>G homozygotes were found to develop a more severe phenotype than c.1187G>A homozygotes or c.536A>G/c.1187G>A compound heterozygotes. All c.536A>G homozygotes developed polyposis and subsequent colorectal malignancy. Comparatively, the cancers occurring in c.1187G>A homozygotes were limited to a single sibship (n=3), with one further c.1187G>A homozygote in the same family developing only a single benign polyp. One of three c.1187G>A/c.536A>G compound heterozygote developed early onset colon cancer. This finding has been previously documented7,13 and is attributed to different effects of these mutations at a functional level. The c.1187G>A mutation acts to structurally modify a C-terminal turn, resulting in partially deficient DNA glycosylase activity, and appears to be less catalytically compromised than the c.536A>G mutation in the N-terminal catalytic base excision domain which results in dramatically reduced glycosylase activity and reduced base-flipping ability2,7,14. The c.536A>G mutation is also more often associated with larger accumulation of endogenous DNA damage 14. Interestingly, in our series, four patients developed malignancy without prior polyposis, of whom three were c.1187G>A homozygotes, and one c.1187G>A/c.933+3A>C compound heterozygote. This is in-keeping with previous literature, highlighting a risk of colorectal cancer in MAP even in the absence of polyposis 15.
Other mutations identified in our cohort included a Portuguese founder mutation, c.1227_1228dupGG (p.Gly410Glyfs) 16 in a Portuguese patient; c.1435G>T (p.Val493Phe), previously reported in a French cohort 17, and c.1147delC (p.Ala385Profs*23), common in patients of Northern European origin 7; and c.933+3A>C (p.Gly264Trpfs*7), which has been reported as an Italian founder mutation, and with lesser frequency in other Western European populations 17, 18. A novel c.337T>C (p.Trp113Arg) variant was detected in two compound heterozygotes with convincing MAP phenotypes. In silico analyses suggest high likelihood of pathogenicity.
A single patient in our cohort with a c.933+3A>C homozygous genotype had the most severe polyp burden in the cohort, as well as early onset malignancy, This mutation alters the sequence of the donor splice site in intron 10, resulting in skipping of exon 10 18. In lymphoblastoid cell lines (LCLs), this splice-site mutation was found to encode a non-functional protein (G264WfsX7). The amount of normal protein produced by LCLs with c.933+3A>C depends on the second mutation, and ranges between 1-100% of that produced by wild type LCLs. In c.933+3A>C/c.933+3A>C homozygous LCLs some primary transcripts were noted to avoid abnormal splicing, resulting in formation of some full-length, wild-type protein 19. Homozygous c.933+3A>C/c.933+3A>C cell lines were noted to produce ~10% normal protein 18. Maintenance of some residual MUTYH activity should predict an attenuated phenotype in c.933+3A>c/c.933+3A>C homozygotes compared to carriers of mutations associated with complete MUTYH inactivation 18. The severe phenotype in the patient with this genotype in our cohort is therefore suggestive of a possible concomitant genetic or environmental modifier.
The risk of colorectal cancer in heterozygous carriers of a single MUTYH mutation is unclear. A large scale meta-analysis suggests that there may be a marginally risk associated with mono-allelic c.536A>G mutations, but not for c.1187G>A, but this effect did not retain statistical significance after Bonferroni correction for multiple analyses 13, and was not felt to be clinically actionable. In this series, no mono-allelic carriers picked up on cascade testing had a history of malignancy or polyposis, and colonoscopic screening undertaken by some of these patients (n=8) revealed a solitary polyp in only one individual. Considering only parents of bi-allelic carriers, this series includes at least 34 obligate carriers, and most did not have a positive history of malignant disease, although cancers were reported in 6 individuals.
Given the phenotype associated with bi-allelic MUTYH mutations, it is strongly recommended that bi-allelic mutation carriers be appropriately identified to facilitate surveillance for colorectal disease. For full siblings of patients with MAP, assuming the parents are obligate carriers, the risk that they are also affected is 1 in 4 (25%), while the risk that they are a mono-allelic carrier is 2/3 (66%). The risk of transmission of a single mutation in MUTYH to the child of a bi-allelic mutation carrier is 100%, but the risk of the child being affected by MAP (i.e. inheriting second mutation from the other parent) depends on the carrier status of the other parent, who has a general population risk (~1%). Cascade testing in our centre is offered to sibs, partners and/or offspring of bi-allelic mutation carriers. Current screening guidelines for mono- and bi-allelic MUTYH mutation carriers are based on recommendations following from a meeting of European experts in Mallorca in 2007 20. Currently, colorectal surveillance of bi-allelic MUTYH mutation carriers follows that of patients with APC-related familial adenomatous polyposis, with colonoscopy recommended to start between 18-20 years, at 2-yearly intervals. Screening of mono-allelic MUTYH-carriers is not recommended outside of national screening programs 20.
The prevalence of bi-allelic MUTYH mutations is expected to be between 1/40,000-1/20,00021. Given a population of ~4.8 million in Ireland, we can estimate that there are 120-240 bi-allelic mutation carriers in the country. A minority of these will be children or young adults who have not yet developed symptoms of the condition. It is clear however, that MUTYH-associated polyposis is under-recognised and under-diagnosed in Ireland. MUTYH-gene analysis should therefore be routinely considered in all patients with polyposis if APC-gene analysis is negative or family history suggests recessive inheritance.
Acknowledgement
The authors would like to thank Ms Amy Nolan (St James’s University Hospital) for her help in retrieving patient demographic information. Dr Terri McVeigh is supported in her work by the HSE/HRB National Academic SpR Fellowship.
Conflict of Interest:
The authors confirm there is no conflict of interest
Correspondence: Terri McVeigh, Department of Clinical Genetics, Our Lady’s Children’s, Hospital Crumlin, Cooley Rd., Dublin 12
Email: [email protected]
Phone : (+353) 01 409 6739
References
1. Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nature reviews Cancer. 2015;15:181-94
2. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nature genetics. 2002;30:227-32.
3. Vogt S, Jones N, Christian D, Engel C, Nielsen M, Kaufmann A, Steinke V, Vasen HF, Propping P, Sampson JR, Hes FJ, Aretz S. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009;137:1976-85.e1-10.
4. Ashton KA, Proietto A, Otton G, Symonds I, Scott RJ. Genetic variants in MUTYH are not associated with endometrial cancer risk. Hereditary cancer in clinical practice. 2009;7:3.
5. Beiner ME, Zhang WW, Zhang S, Gallinger S, Sun P, Narod SA. Mutations of the MYH gene do not substantially contribute to the risk of breast cancer. Breast cancer research and treatment. 2009;114:575-8.
6. LOVD Homepage, Leiden University Medical Center, http://chromium.lovd.nl/LOVD2/colon_cancer/home.php?select_db=MUTYH
7. Nielsen M, Joerink-van de Beld MC, Jones N, Vogt S, Tops CM, Vasen HF, Sampson JR, Aretz S, Hes FJ. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology. 2009;136:471-6
8. Miyaki M, Iijima T, Yamaguchi T, Hishima T, Tamura K, Utsunomiya J, Mori T. Germline mutations of the MYH gene in Japanese patients with multiple colorectal adenomas. Mutation research. 2005;578:430-3.
9. Yamaguchi S, Ogata H, Katsumata D, Nakajima M, Fujii T, Tsutsumi S, Asao T, Sasaki K, Kuwano H, Kato H. MUTYH-associated colorectal cancer and adenomatous polyposis. Surgery today. 2014;44:593-600.
10. Lejbkowicz F, Cohen I, Barnett-Griness O, Pinchev M, Poynter J, Gruber SB, Rennert G. Common MUTYH mutations and colorectal cancer risk in multiethnic populations. Familial cancer. 2012;11:329-35
11. National Cancer Registry of Ireland, http://www.ncri.ie/ (accessed 25/11/2015)
12. Cressey D. DNA repair sleuths win chemistry Nobel. Nature. 2015;526(7573):307-8.
13. Theodoratou E, Campbell H, Tenesa A, Houlston R, Webb E, Lubbe S, Broderick P, Gallinger S, Croitoru EM, Jenkins MA, Win AK, Cleary SP, Koessler T,Pharoah PD, Küry S, Bézieau S, Buecher B, Ellis NA, Peterlongo P, Offit K, Aaltonen LA, Enholm S, Lindblom A, Zhou XL, Tomlinson IP, Moreno V, Blanco I,Capellà G, Barnetson R, Porteous ME, Dunlop MG, Farrington SM. A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. British journal of cancer. 2010;103:1875-84.
14. Molatore S, Russo MT, D'Agostino VG, Barone F, Matsumoto Y, Albertini AM, Minoprio A, Degan P, Mazzei F, Bignami M, Ranzani GN. MUTYH mutations associated with familial adenomatous polyposis: functional characterization by a mammalian cell-based assay. Human mutation. 2010;31:159-66.
15. Cleary SP, Cotterchio M, Jenkins MA, Kim H, Bristow R, Green R, Haile R, Hopper JL, LeMarchand L, Lindor N, Parfrey P, Potter J, Younghusband B, Gallinger S. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009;136:1251-60.
16. Isidro G, Laranjeira F, Pires A, Leite J, Regateiro F, Castro e Sousa F, Soares J, Castro C, Giria J, Brito MJ, Medeira A, Teixeira R, Morna H, Gaspar I, Marinho C, Jorge R, Brehm A, Ramos JS, Boavida MG. Germline MUTYH (MYH) mutations in Portuguese individuals with multiple colorectal adenomas. Human mutation. 2004;24:353-4.
17. Olschwang S, Blanche H, de Moncuit C, Thomas G. Similar colorectal cancer risk in patients with monoallelic and biallelic mutations in the MYH gene identified in a population with adenomatous polyposis. Genetic testing. 2007;11:315-20.
18. Pin E, Pastrello C, Tricarico R, Papi L, Quaia M, Fornasarig M, Carnevali I, Oliani C, Fornasin A, Agostini M, Maestro R, Barana D, Aretz S, Genuardi M, Viel A. MUTYH c.933+3A>C, associated with a severely impaired gene expression, is the first Italian founder mutation in MUTYH-Associated Polyposis. International journal of cancer. 2013;132:1060-9.
19. Mazzei F, Viel A, Bignami M. Role of MUTYH in human cancer. Mutation research. 2013;743-744:33-43.
20. Vasen HF, Moslein G, Alonso A, Aretz S, Bernstein I, Bertario L, Blanco I, Bülow S, Burn J, Capella G, Colas C, Engel C, Frayling I, Friedl W, Hes FJ, Hodgson S, Järvinen H, Mecklin JP, Møller P, Myrhøi T, Nagengast FM, Parc Y, Phillips R, Clark SK, de Leon MP, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Tejpar S, Thomas HJ, Wijnen J. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut. 2008;57:704-13.
21. Aretz S, Genuardi M, Hes FJ. Clinical utility gene card for: MUTYH-associated polyposis (MAP), autosomal recessive colorectal adenomatous polyposis, multiple colorectal adenomas, multiple adenomatous polyps (MAP) - update 2012. European journal of human genetics : EJHG. 2013;21(1)
P485