Introduction
FXIII is a pro-coagulant plasma protein that promotes clot stability by cross-linking fibrin strands. First reported in 19601, congenital severe FXIII deficiency (<1% of normal, 0.01IU/mL) causes a life-long bleeding tendency classically associated with neonatal umbilical stump bleeding. Acquired FXIII deficiency typically occurs due to the development of inhibitory autoantibodies, presenting in older individuals with no preceding personal or family bleeding history. FXIII-inhibitors may be spontaneous or associated with medications (most commonly isoniazid and phenytoin), autoimmune disorders or malignancy. Establishing the diagnosis proves challenging, as routine coagulation screening tests (PT, APTT, fibrinogen) are normal. Consequently, FXIII deficiency requires a high index of clinical suspicion and FXIII testing, normally performed in a Haemostasis Reference Laboratory. Critically, identifying FXIII deficiency has direct clinical importance, as specific replacement therapy with FXIII concentrate is required to arrest bleeding in these patients. In contrast, standard transfusion approaches will not be sufficient to control ongoing or life-threatening bleeding. In order to increase awareness of FXIII deficiency in patients with unexplained significant bleeding, we present a case of idiopathic acquired FXIII deficiency and discuss optimal clinical management.
Case Report
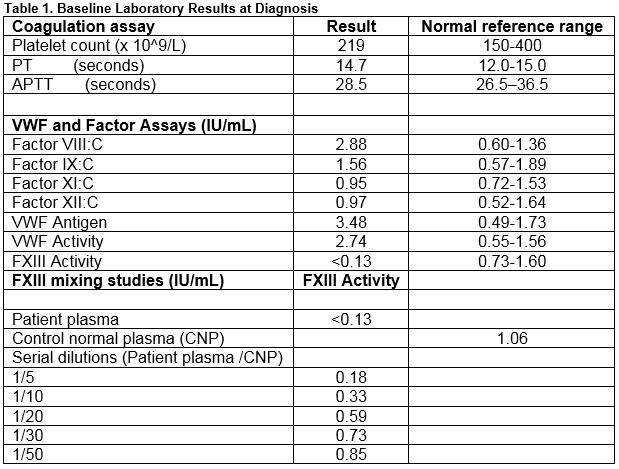
A 70-year-old man presented with spontaneous, extensive left gluteal bruising with a large haematoma confirmed on ultrasound. He had no personal or family history of bleeding. Laboratory analysis demonstrated normal PT, APTT and fibrinogen. Given the unusual bleeding pattern, an acquired bleeding disorder was suspected. Coagulation factors (Factors II, V, VII, X, VIII, IX, IX and XII) were normal and Von Willebrand Factor levels were mildly elevated, consistent with acute phase response (Table 1). We proceeded to test FXIII levels and found markedly reduced plasma FXIII activity levels (<0.13 IU/mL; normal reference range 0.73 IU/mL-1.60IU/mL, Berichrom FXIII activity assay, Dade Berichrom).
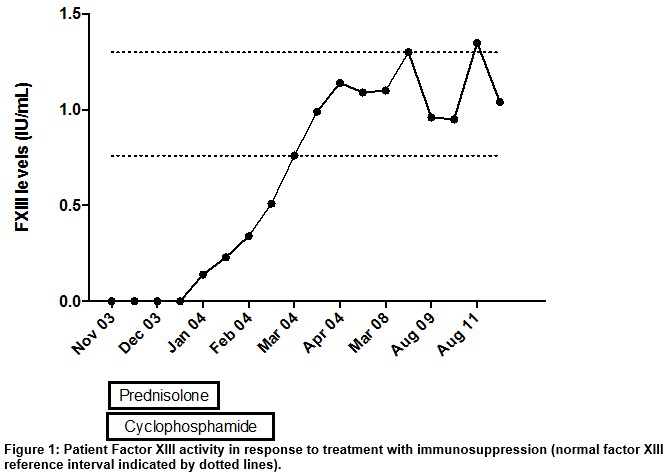
To understand the pathophysiology underlying this reduction in plasma FXIII levels, we performed standard plasma mixing studies (Table 1), which confirmed the presence of a neutralizing inhibitory FXIII autoantibody. Consequently, FXIII-inhibitor eradication was commenced using immunosuppression with intravenous immunoglobulin (IVIg) (1g/kg for two days), cyclophosphamide 1.5mg/kg and prednisolone 1mg/kg daily. No further bleeding complications occurred and FXIII replacement was not required. The gluteal haematoma resolved over the following four weeks. Despite extensive investigations, no underlying precipitant was identified. FXIII levels normalised over the following four months and immunosuppression subsequently weaned (Figure 1). There is no evidence of relapse at long-term follow up (13 years).
Discussion
Acquired FXIII deficiency is a rare disorder with only thirty-eight cases reported to date2–4. Acquired FXIII deficiency usually occurs in patients >60 years and may present with severe, spontaneous bleeding, including fatal intracranial haemorrhage2. The diagnosis is easily overlooked and commonly involves a significant time-delay. Critically, arriving at the correct diagnosis is dependent upon a high index of suspicion coupled with specialised laboratory testing5.
Given the rarity of the condition, defining optimal treatment for acquired FXIII deficiency can be particularly challenging6. Management requires expert care in a Haemophilia Comprehensive Care setting, acute treatment of haemorrhage and inhibitor eradication. Although fresh frozen plasma contains FXIII, plasma transfusion alone will not achieve haemostatic FXIII levels. Consequently, specific FXIII concentrates are required, often in high doses (50-150 IU/kg). Furthermore, due to the FXIII-inhibitor, the half-life of the infused FXIII replacement may be significantly shortened. Thus, alternate adjunctive treatments may be considered, particularly in refractory bleeding including plasma exchange or cryoprecipitate(7). Definitive treatment requires eradication of the FXIII-inhibitor, usually involving a significant lag period during which the risk of bleeding remains high2 (Figure 1). In the interim, patients require close monitoring and aggressive treatment of haemorrhage.
Contributions:
H.F. & M.L. reviewed the clinical data and prepared the manuscript, all authors (H.F., M.B., N.M.O.C, J.O.D., B.W. & M.L.) were involved in reviewing the manuscript.
Declaration of Conflicts of Interest:
The authors declare that there is no conflict of interest regarding the publication of this article.
Correspondence
Dr. Michelle Lavin, National Coagulation Centre, St James’s Hospital, Dublin, Ireland.
Tel +353 (1) 416 2141
Email: [email protected]
References:
1. Duckert F, Jung E, Shmerling DH. A hitherto undescribed congenital haemorrhagic diathesis probably due to fibrin stabilizing factor deficiency. Thromb Diath Haemorrh 1960 Dec 15;5:179–86.
2. Kessel R, Hu C, Shore-Lesserson L, Rand J, Manwani D. A child with acquired factor XIII deficiency: case report and literature review. Haemophilia 2013 Nov [cited 2014 Nov 11];19(6):814–26.
3. Biswas A, Ivaskevicius V, Thomas A, Oldenburg J. Coagulation factor XIII deficiency. Diagnosis, prevalence and management of inherited and acquired forms. Hamostaseologie 2014 Jan;34(2):160–6.
4. Tha MH, Tien SL. Acquired factor XIII deficiency: still a clinical challenge in the era of novel therapy. Haemophilia 2014 Jan;20(1):e104-5.
5. Kohler HP, Ichinose A, Seitz R, Ariens RAS, Musbek L. Diagnosis and classification of factor XIII deficiencies. J Thromb Haemost 2011 Jul;9(7):1404–6.
6.Boehlen F, Casini A, Chizzolini C, Mansouri B, Kohler HP, Schroeder V, Reber G, de Moerloose P. Acquired factor XIII deficiency: a therapeutic challenge. Thromb Haemost 2013;109(3):479–87.
7. Collins PW, Chalmers E, Hart D, Jennings I, Liesner R, Rangarajan S, Talks K, Williams M, Hay CRM. Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO. Br J Haematol 2013 Sep;162(6):758–73.
(P757)