Introduction
Hyperammonaemia is a pathological accumulation of ammonia in the blood. Under physiological circumstances, low concentrations of ammonia are detectable. Normal ranges of ammonia in a neonate are less than 100µmol/L and in a child less than 50 µmol/L 1. Ammonia is neurotoxic and is therefore rapidly detoxified, mainly by the formation of non-toxic urea via the urea cycle and also by glutamine synthesis 2.
Hyperammonaemia can be primary or secondary. Primary causes involve inborn metabolic disorders affecting the urea cycle. Secondary hyperammonaemia can be caused by toxic metabolites or drugs interfering with the function of the urea cycle or liver disease 3. Hyperammonaemia has potentially life-threatening complications; symptoms are essentially neurological in nature. The mechanisms through which ammonia leads to brain damage are complex. The presentation of hyperammonaemia may vary with age as some effects are specific to the developing brain 4.
Hyperammonaemia can present at any age, depending on the underlying cause and its severity. In the neonatal period hyperammonaemia typically presents after a short symptom-free interval; symptoms include poor feeding, vomiting, lethargy, irritability and tachypnoea; deterioration may progress rapidly leading to encephalopathy and coma; it may also present as sepsis-like neonatal crisis. Prognosis depends on ammonia peak concentrations and duration of the hyperammonaemic status 1. In infants and young children symptoms are, typically, less acute and more variable; these consist of episodic vomiting, failure to thrive, lethargy or developmental delay. In older children and adolescents hyperammonaemia may present acutely and precipitated by a metabolic stress such as illness, prolonged fasting or high protein intake; it may also present chronically with neurological or psychiatric symptoms. At any age, acute hyperammonaemia may lead to an altered level of consciousness and may mimic encephalitis or drug intoxication 5. The mortality rate depends on the underlying aetiology, its severity and commencement of and response to treatment 3. Ammonia neurotoxicity becomes irreversible after a short interval. As the clinical outcome is strongly influenced by the severity and the duration of the hyperammonaemic state, immediate treatment is essential 1. We retrospectively reviewed our computerized database at the National Centre for Inherited Metabolic Disorders with approval/consent as appropriate. We present clinical presentation and diagnostic approaches in a paediatric case series of patients with hyperammonaemia and encephalopathy due to an inborn metabolic disorder.
Clinical Study
Case 1
A 9-day old girl presented with lethargy and poor feeding with no relevant background history. Hypothermia and bradycardia were noted. She was pale with poor perfusion with a reduced Glasgow Coma Scale (GCS) of 8/15. Abdominal examination revealed mild hepatomegaly. She was resuscitated and commenced on intravenous fluids and antibiotics. Blood tests revealed respiratory alkalosis, undetectable urea (normal range (NR) 1.0-6.0mmol/L) and very high ammonia level of 562µmol/L. She was subsequently transferred to the Paediatric Intensive Care Unit (PICU) for further management with a working diagnosis of urea cycle defect for which she was started on sodium benzoate, arginine and sodium phenylbutyrate to induce alternative pathways of nitrogen removal. Plasma amino acids revealed elevated glutamine (2213µmol/L, NR 475-960), citrulline (565µmol/L, NR 8-63) and argininosuccinate (1014µmol/L, NR – undetectable) with its anhydrides. This was consistent with Argininosuccinate Lyase Deficiency, a primary urea cycle defect, which was confirmed at a molecular genetic level. She responded well to treatment and was discharged home with regular follow-up appointments.
Case 2
An 8-month old baby girl presented with poor intake associated with gastroenteritis. On examination she was unresponsive, pale, dehydrated and had moderate hepatomegaly. She was resuscitated with intravenous fluids and dextrose. Initial blood tests revealed extremely low blood glucose (0.06mmol/L) with low ketones (0.65mmol/L). Blood gas analysis revealed metabolic acidosis with a pH of 7.22 (NR 7.35-7.45) with normal anion gap and lactate. Liver function tests were elevated (AST (aspartate aminotransferase) 777U/L (NR 0-45) and ALT (alanine aminotransferase) 1191U/L (NR 0-65)). Ammonia was raised at 172µmol/L and CK (creatine kinase) was elevated at 369U/L (NR 20-155). Urine organic acids revealed dicarboxylic aciduria with low ketones along with hexanoylglycine, suberylglycine and phenylpropionylglycine excretion. Blood acylcarnitines showed elevated hexanoylcarnitine (C6 carnitine, 0.27µmol/L, NR < 0.12), octanoylcarnitine (C8 carnitine 3.6µmol/L, NR < 0.22) and decenoylcarnitine (C10:1 carnitine 0.46µmol/L, NR <0.25) with low free carnitine in blood at 3.5µmol/L (NR 15.5-46.7). Results were consistent with MCAD (Medium Chain Acyl-CoA Dehydrogenase) deficiency. She was started on regular feeds, oral carnitine, restricted fasting times and carbohydrate supplements when unwell. Fibroblast studies and molecular genetic analysis confirmed the diagnosis.
Case 3
A 2-day old boy presented with poor feeding and high-pitched cry. Birth history was uneventful. On examination he was lethargic, poorly perfused and in respiratory distress. He was resuscitated with intravenous fluids containing dextrose and further support was provided by intubation and ventilation. His blood glucose was reduced at 2.1mmol/L along with ketonaemia/ketonuria. Blood gas revealed metabolic acidosis with a pH of 7.2, pCO2 of 4.1, base excess of -7.2 and bicarbonate of 18.2. Anion gap was raised at 24.8mmol/L (NR 10-20). Ammonia was high at 583µmol/L. Plasma amino acids showed elevated glycine (765µmol/L, NR <515). Urine organic acids were abnormal with 3-hydroxypropionate, tiglylglycine, methylcitrate and propionylglycine. Acylcarnitine profile showed marked increase in propionylcarnitine (30.2µmol/L, NR 0.0-6.18). These findings were consistent with the diagnosis of Propionic acidaemia which was confirmed at an enzyme/molecular genetic level. He responded well to treatment consisting of a low protein diet with the addition of synthetic protein which is methionine, threonine and valine free and low in isoleucine and medication with carnitine, biotin and metronidazole.
Discussion
We present diagnostic approaches and clinical courses in a paediatric case series of patients with hyperammonaemia and encephalopathy with rare inborn metabolic disorders. Our aim is to raise the awareness of early detection, emergency treatment and immediate blood and urine to investigate for inborn metabolic disorders to tailor treatment in affected individuals with a view to improving the overall outcome.
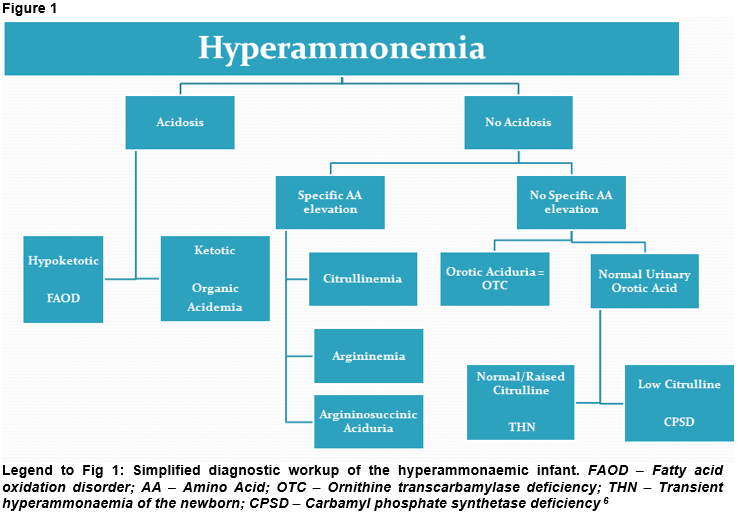
Workup for hyperammonaemia includes, e.g., blood ammonia, blood gases, glucose, lactate, urea and electrolytes. Ketones should be measured in blood and urine. Along with these, basic laboratory investigations are key. The presence of acidosis with a raised anion gap or ketosis/ketonuria in the setting of hyperammonaemia can point towards organic acidaemias (Fig 1) which are disorders caused by a defect in intermediary metabolism of, e.g., amino acids and fatty acids along with the accumulation of toxic organic acids. Hyperammonaemia with acidosis but low ketones may be caused by fatty acid oxidation defects (Fig 1) such as MCAD deficiency or long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD-D). These conditions can lead to hypoketotic hypoglycaemia particularly during intercurrent illnesses.
Hyperammonaemia caused by an enzyme deficiency in the urea cycle may be characterized by respiratory alkalosis and a low pCO2 (Fig 1), however this can be masked by apnoea and hypoxia due to lactic acidosis. It is important at any stage to ensure adequate oxygenation and perfusion 6. Hyperammonaemia resulting in cerebral oedema is typically preceded by hyperventilation along with respiratory alkalosis which may be caused by direct toxicity 7.
If hyperammonaemia is confirmed, further specialist metabolic investigations to reach a conclusive diagnosis are necessary which include: quantitative amino acids in blood and urine, organic acid analysis in urine, acylcarnitine profile in blood. As prognosis and neurological outcome are strongly associated with the magnitude and duration of the hyperammonaemia, treatment should not be delayed 3. Initial management comprises of stopping protein intake temporarily and fluid resuscitation with dextrose and electrolytes to provide hydration and promote anabolism. General supportive care must be added as clinically warranted. Hyperammonaemia in symptomatic individuals usually requires the use of medications which serve as nitrogen scavengers or substrates that may be deficient. Patients with encephalopathy due to severe hyperammonaemia (i.e., > 500 μmol/L) may require dialysis for detoxification in addition to conservative treatment1. Long-term treatment often involves a specialized diet and monitoring. Early implementation of ‘unwell day’/emergency regimes is mandatory and long-term multi-disciplinary team input is crucial.
Hyperammonaemia is a medical emergency; if untreated it can progress to severe encephalopathy, coma and death; surviving patients often have neurological sequelae. There should be a low threshold for suspicion of hyperammonaemia in neonates with neurological deterioration after a normal initial adaptation. Initial treatment, stabilization or resuscitation and standard management should not be delayed. In any patients presenting with a suspicion of hyperammonaemia, plasma ammonia should be measured without delay. In a patient with hyperammonaemia, an inborn metabolic disorder affecting the capacity of the urea cycle should be considered and appropriate investigations performed.
Conflicts Of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper. We obtained ethical/research approval in writing from our institutional Ethics and Research Committee and informed consent as appropriate.
Corresponding Author
Sheiniz Giva,
National Centre for Inherited Metabolic Disorders,
Temple Street Children’s University Hospital,
Dublin 1,
Ireland
Email: [email protected]
References
1. Unsinn C., Das A., Valayannopoulos V., Thimm E., Beblo S., Burlina A., Konstantopoulou V., Mayorandan S., de Lonlay P., Rennecke J., Derbinski J., Hoffmann G. F. and Häberle J. Clinical Course of 63 Patients with Neonatal Onset Urea Cycle Disorders in the Years 2001-2013. Orphanet J Rare Dis (2016); 11:116. 2. Guder W. G., Häussinger D. and Gerok W. Renal and hepatic nitrogen metabolism in systemic acid base regulation. J Clin Chem Clin Biochem 1987;25:457-66. 3. Häberle J. Clinical and Biochemical Aspects of Primary and Secondary Hyperammonemic Disorders. Arch Biochem Biophys (2013) 15; 536: 101-8. 4. Braissant O., McLin V. A. and Cudalbu C. Ammonia Toxicity to the Brain. J Inherit Metab Dis (2013) 36:595-612. 5. Nassogne M. C., Héron B., Touati G., Rabier D. and Saudubray J. M. Urea Cycle Defects: Management and Outcome. J Inherit. Metab Dis (2005); 28:407-414. 6. Nyhan W. L., Barshop B. A. and Ozand P. T. Atlas of Metabolic Diseases. 2nd ed. London: Hodder Arnold, 2005. 7. Adeva M. M., Souto G., Blanco N. and Donapetry C. Ammonium metabolism in humans. Metabolism 2012;61:1495-511.
P858