Introduction
MELAS is a progressive neurodegenerative disorder affecting multiple organ systems, especially those with high energy oxidation metabolism like muscle, brain, eye and heart. It is due to a point mutation in mitochondrial DNA which is maternally inherited1. Presenting features include developmental delay and stroke-like episodes. Other features include hearing loss, cardiomyopathy, short stature and fatigue2.There is no curative treatment. The disease remains progressive and fatal.
Case 1
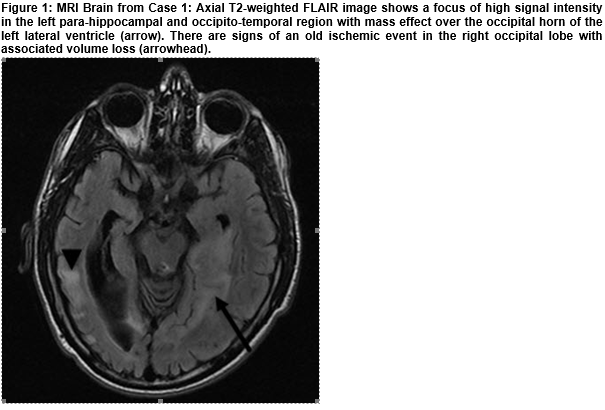
An ex-premature male, born at 30 weeks gestation. At age 14 months, he had global developmental delay and bilateral sensorineural hearing loss, but was attending mainstream school with a special needs assistant. Aged 16, he presented with sudden onset confusion, headache, left sided weakness and visual loss. He regained some vision and use of his left side and was discharged. One week later he presented with right sided weakness and was re-admitted. He had 3 seizures on the night of his admission. MRI showed chronic and acute CVA (Figure 1). The combination of learning difficulties, deafness and stroke-like episodes made MELAS likely. Blood and urine DNA samples sent for the m.3243A>G (MELAS) mutation identified the mutation at a level of 59% heteroplasmy in blood and 95% heteroplasmy in urine. Unfortunately he exhibits significant residual dysphasia and reduced cognition and is unable to continue at mainstream school.
Case 2
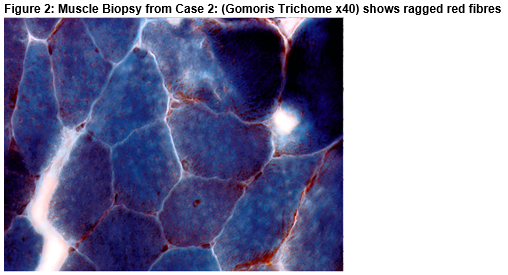
A 3-year-old male with global developmental delay. At age 9, he presented with drowsiness and left sided hemiplegia. Three months later, he presented with drowsiness and right-sided hemiplegia. A presumed diagnosis of subclinical status epilepticus was made. He was administered intravenous midazolam and subsequently had a respiratory arrest. He was presumed to have seizures and commenced on Levetiracetam. Two months later he re-presented with headache and left-sided hemiplegia and administered intravenous Lorazepam which resulted in respiratory arrest. The following month he had tonic-clonic seizures, was administered Diazepam per rectum and had a third respiratory arrest requiring his third intubation, ventilation and transfer to PICU. At this time his venous blood gas showed lactic acidosis (pH 7.16, Lactate10.3). The combination of encephalopathy, lactic acidosis and stroke-like episodes made the diagnosis of MELAS likely. Muscle biopsy showed ragged red fibres(Figure 2).
Discussion
Mitochondrial disorders comprise a heterogeneous group of disorders, of which MELAS is rare. The most common disease-causing mtDA mutation in MELAS is m.3243A>G, found in80% of patients3 as in case 1. Recent population-based studies suggest that the carrier rate of this mutation is 1 in 4004.
Both patients developed stroke-like episodes, a hallmark of MELAS. These are thought to represent metabolic strokes due to deficiency of adenosine triphosphatase, increased lactic acid production, mitochondrial angiopathy, or changes in nitric oxide metabolism5. Case 2 was initially diagnosed with epilepsy, which is strongly associated with stroke-like episodes, though sodium valproate is contraindicated in mitochondrial disorders as it exacerbates seizures6. It is important that the distinction between seizures and stroke is made: case 2 attended three times with alternating hemiplegia, and as a result of benzodiazepine administration, suffered 3 respiratory arrests. This extreme reaction to benzodiazepines is rare, and not reported in the literature.
In addition to stroke-like episodes, both cases had a background of developmental delay and macrocephaly, which were assigned alternate diagnoses. In the largest cohort study of MELAS patients with the m.3243A>G mutation to date, the authors suggested that diagnoses could be considered in patients with 3 or more of the following: developmental delay, deafness, cardiomyopathy, epilepsy, migraine, diabetes mellitus, gastrointestinal disturbance and retinopathy7. L-Arginine during stroke-like episodes can reduce cerebral damage8. Algorithms for diagnosis in paediatric patients have been suggested9. It is important to reach a diagnosis early as juvenile-onset cases can rapidly decline and have earlier mortality(median age from onset to death 6.4years in juvenile compared with 10.2years in adult onset)10. Repeated seizures cause ventricular dilatation, cerebral atrophy and basal ganglia calcification. There is cognitive decline and debilitating myopathy. The clinical course can rapidly decline, leading to profound disability and premature death. Early consideration of MELAS in a patient with stroke can reduce morbidity and improve quality of life.
Correspondence: N Quinn
Emergency Department, Royal Children's Hospital, Melbourne, Australia
Email: [email protected]
Conflict of Interest
The authors do not have any conflict of interest to disclose
Acknowledgements
J Grindlay, PEM Consultant at RCH
Referenc
1. Kamal-Salah R, Baquero-Aranda I, Grana-Perez M, Garcia-Campos J. Macular pattern dystrophy and homonymous hemianopia in MELAS syndrome. BMJ Case Reports 2015; doi:10.1136/bcr-2014-206499
2. Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis and stroke-like episodes: basic concepts, clinical phenotype and therapeutic management of MELAS syndrome. Ann N Y Acad Sci 2008;1142:133-58
3. Vanniarajan A, Nayak D, Reddy AG, Singh L, Thangaraj K. Clinical and genetic uniqueness in an individual with MELAS. Am J Med Genet B Neuropsychiatr Genet 2006;141B:440–4
4. Aurangzeb S, Vale T, Tofaris G, Poulton J, Turner M. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) in the older adult. Pract Neurol 2014;14:432-436
5. Kimata KG.; Gordan L, Ajax ET, Davis PH, Grabowski T, A Case of Late-Onset MELAS. Arch Neurol 1998;55:722-5.
6. Lam CW1, Lau CH, Williams JC, Chan YW, Wong LJ., Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) triggered by valproate therapy. Eur J Pediatr 1997;156:562–4
7. Nesbitt V1, Pitceathly RD, Turnbull DM, Taylor RW, Sweeney MG, Mudanohwo EE, Rahman S, Hanna MG, McFarland R.,The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation—implications for diagnosis and management. J Neurol Neurosurg Psychiatry 2013;84:936-8
8. Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, Fujimoto S, Matsuishi T. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology 2005;64:710-712
9. Wolf NI, Smeitink JA. Mitochondrial disorders: A proposal for consensus diagnostic criteria in infants and children. Neurology 2002;59:1402-1405
10. Goodfellow JA, Dani K, Stewart W, Santosh C, McLean J, Mulhern S, Razvi S. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes: an important cause of stroke in young people. Postgrad Med J 2012;88:326–34
p455