An Approach to Hypophosphataemia
J Martin-Grace, M J. McKenna
Dept of Endocrinology, St. Vincent’s University Hospital, Elm Park, Dublin 4
Phosphate homeostasis
Phosphate is a predominantly intracellular anion, 85% of which is stored in bone as hydroxyapatite crystals and calcium phosphate, providing strength and rigidity to bone. Approximately 15% is present intracellularly as phosphate ions, esters and phosphoproteins, and is involved in post receptor signalling, translation of DNA and RNA, and acid-base buffering, with the remainder present in extracellular fluid1. The physiological control of phosphate is chiefly under the regulation of activated vitamin D (calcitriol), parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23), which act by modifying uptake from dietary sources, renal reabsorption and excretion, and release from skeletal stores. Hypophosphataemia results from three main mechanisms, increased renal excretion (PTH excess, FGF23 excess, and renal tubular disorders such as Fanconi syndrome), decreased gastrointestinal absorption and exchange from extra- to intra-cellular compartments2.
Pathogenesis
Gastrointestinal absorption
The majority of dietary phosphorus is absorbed in the jejunum. Jejunal absorption is achieved by both sodium dependent and independent transport. Sodium-dependent active transportation regulated by both dietary phosphate load and calcitriol, although oestrogens, glucocorticoids and metabolic acidosis are all thought to play a part. PTH indirectly influences dietary absorption by stimulating the renal production of calcitriol, which in turn increases absorption of ingested phosphate alongside calcium. The absorbed phosphate enters the extracellular fluid and is exchanged into the bone and intracellular stores as required. Isolated dietary deficiency causing hypophosphataemia is rare, although it can occur in conjunction with vitamin D deficiency, malnutrition, malabsorptive states, or use of phosphate binders or anti-acid medication.
Renal phosphate homeostasis
Phosphate is freely filtered at the level of the nephron, and excretion is therefore determined by the rate of proximal tubule resorption. Resorption is inhibited by both PTH and FGF-232. PTH secretion is stimulated by reduction in calcium concentration. In addition to activation of calcitriol to increase GI absorption of both calcium and phosphate, PTH will stimulate bone resorption by osteoclasts, releasing both phosphate and calcium from skeletal stores. PTH will also reduce proximal renal tubular resorption of phosphate, and therefore the net effect is a rise in serum calcium concentration and decrease in serum phosphate. PTH secretion is inhibited by both FGF-23 and by negative feedback from calcitriol. FGF-23, the most widely studied phosphatonin, is predominantly expressed by bone. In conjunction with its co-receptor klotho, it reduces phosphate reabsorption from the proximal renal tubules by downregulation expression of the sodium/inorganic phosphate transported on the apical membrane of proximal renal tubular cells. Conditions that increase expression of FGF23, either due to genetic mutations such as occurs in X-linked hypophosphataemia (XLH) and autosomal dominant hypophosphataemic rickets (ADHR), or due to increased FGF 23 production by activated osteoblasts in fibrous dysplasia/ Mc Cune-Albright syndrome(MAS), cause a phosphaturia resulting in hypophosphataemia .
Cellular exchange
Acute hypophosphataemia can occur in the setting of refeeding syndrome. During the preceding period of poor nutrition, whole body phosphate stores are depleted, although serum phosphate may be normal. Once feeding restarts, cellular uptake of phosphate increases, causing hypophosphataemia, which must be monitored for and replaced as required. Diabetic ketoacidosis can also induce transient hypophosphataemia, as the osmotic diuresis induced by hyperglycaemia depletes phosphate stores, and re-introduction of insulin causes increased cellular uptake of phosphate, but replacement of phosphate in this particular context does not improve clinical outcomes, and may worsen hypocalcaemia3. Respiratory alkalosis resulting from hyperventilation, mechanical ventilation and salicylate poisoning can similarly cause an acute hypophosphataemia as phosphate moves intracellularly during the alkalosis, but normalises with resolution of alkalosis2.
Clinical presentation
Symptoms are typically non-specific if present, and include generalised muscle weakness, fatigue and occasionally myalgia. Hypophosphataemia can impair neuromuscular function, and therefore in the acute severe setting can cause impaired respiratory or cardiac function, and alterations in mental status2. Chronic hypophosphataemia in adults can cause osteomalacia, which results in bone pain both in the presence or absence of associated pseudofractures. In children, chronic hypophosphataemia during periods of growth can cause rickets, and therefore may present with short stature and bowing skeletal deformity2. Evaluation should include a careful family history and clinical examination focusing on weakness, skeletal deformity or features suggestive of rickets, measurement of height and range of movement at the large joints, which may be limited in cases of X-linked hypophosphataemia due to calcification of entheses. The presence of facial deformity, café au lait spots, or in children the presence of precocious puberty should raise suspicion for the presence of Mc Cune-Albright syndrome4.
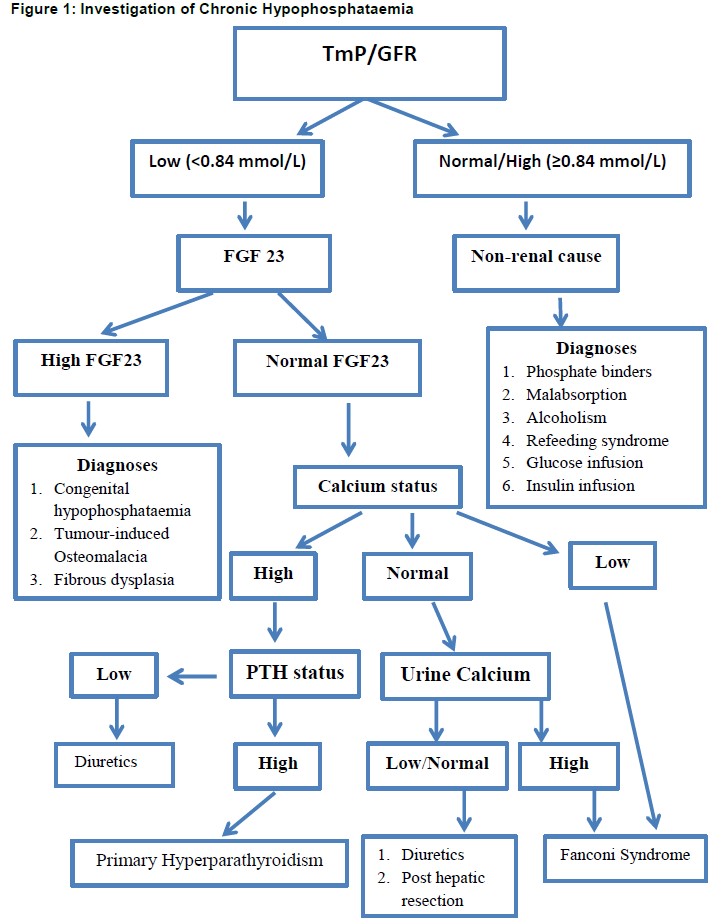
Investigation of chronic hypophosphataemia
Assessment of renal handling of phosphate is an important test in the investigation of chronic hypophosphataemia, as it allows the clinician to distinguish between renal and non-renal pathology .This is achieved by the measurement of renal phosphate excretion, which in the case of renal hypophosphataemia will be inappropriately high. Renal phosphate threshold is assessed by collecting a fasting serum sample and two hour timed urine sample for measurement of phosphate and creatinine in both serum and urine. The TmP/GFR is then derived from a formula. If TmP/GFR is low in the setting of hypophosphataemia, this indicates renal phosphate wasting; whereas a normal or high TmP/GFR implies appropriate phosphate conservation, indicating the explanation for hypophosphataemia is not renal in origin [figure 1].
PTH and calcium should be measured, because hypophosphataemia accompanies hyperparathyroidism, both primary and secondary. The normal physiological response to hypophosphataemia is an increase in calcitriol, but in FGF23-mediated hypophosphataemia renal production of calcitriol is impaired by the direct action of FGF23. Urinary calcium may be raised in hyperparathyroidism, which can arise as a result of FGF23 mediated hypophosphataemia as well as exist as a cause for hypophosphataemia in its own right.
Plain radiographs, in severe cases of hypophosphataemia with osteomalacia, may identify a special type of stress fracture called Looser zones. Plain radiographs should be performed of any areas of deformity in order to assess for ‘ground glass’ appearance suggestive of fibrous dysplasia (FD). Bone scintigraphy is a more sensitive way to identify Looser zones or FD lesions. While bone mineral density on DXA scan is typically reduced in patients with osteomalacia, it can be increased in X-linked hypophophataemia, a condition of FGF23 dependent renal phosphate wasting resulting from an inactivating mutation in the PHEX gene5. Further genetic analysis can be performed in the case of hypophosphataemic rickets for mutations of PHEX, FGF23, DMP1 & ENPP1.
Management
The management of hypophosphataemia depends on the underlying aetiology, and is ideally targeted at the cause. Acute hypophosphataemia in a hospital setting in the context of refeeding syndrome can be managed by regular monitoring of at risk patients, dietician review of at-risk patients, and supplementation as needed. In settings such as respiratory alkalosis and diabetic ketoacidosis, it is self-limiting and resolves as the underlying metabolic disturbance resolves. Intravenous phosphate therapy should be used with caution, and should not be given to patients with co-existent hypocalcaemia. Oral supplementation is preferable in the stable patient. Hypocalcaemia in the presence of hypophosphataemia may suggest an underlying vitamin D deficiency, which can be managed by vitamin D replacement2.
Chronic FGF23 mediated hypophosphataemia is managed with replacement of firstly calcitriol and then phosphate. In adult patients, the goal of therapy is to reduce osteomalacia, and improve fracture healing and bone pain, rather than targeting serum phosphate concentration in the normal range. The dose required for an individual patient will be titrated to serum PTH, calcitriol and both serum and urinary calcium, which will increase with replacement therapy2,6. The emergence of hypercalcaemia or hypercalcuria on treatment should prompt a dose reduction. Anti-resorptive therapy in the form of bisphosphonates may be of benefit in certain patient groups, such as FD4. Patients with tumour induced osteomalacia should have the primary tumour completely resected if possible2.
Conclusion
Hypophosphataemia is common both in an acute and chronic setting, and among its many roles in maintaining homeostasis, is important for both bone growth in a paediatric setting, and bone mineralisation. Its presentation may be non-specific, however treatment can significantly improve the consequences of long term hypophosphataemia. An evaluation of the renal threshold of phosphate is an important investigation in determining the aetiology. FGF-23 independent hypophosphataemia is directed at the cause where possible. Patients with FGF-23 dependent mechanisms require ongoing follow up to facilitate careful monitoring of both serum and urinary calcium and phosphate while on replacement therapy.
Declaration of conflict of interest:
Nil
Correspondence
Professor Malachi J. Mc Kenna, Consultant Endocrinologist St Michael’s Hospital & St Vincent’s University Hospital
References:
1. Goretti M. Phosphate homeostasis and its role in bone health. Pediatr Nephrol (2012) 27:2039–2048.
2. Imel EA, Econs MJ . Approach to the hypophosphataemic patient. J Clin Endocrinol Metab 2012;97:696-706.
3. Fischer JN, Kitabchi AE. A Randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. JCEM 1983;57(1):177-80.
4. McGowan A, McKenna MJ. Fibrous Dysplasia; challenges in diagnosis and treatment. Hospital doctor of Ireland 2016;21(10).
5. Reid IR, Murphy WA, Hardy DC, Teitelbaum SL, BergfieldMA, Whyte MP. X-linked hypophosphataemia: skeletal mass in adults assessed by histomorphometry, computed tomography & absorptiometry. Am J Med 90:63-69.
6. Carpenter TO, Imel EA Holm IA, Jan de Beur SM, Insogna KL. A Clinician’s guide to X linked hypophosphataemia. J Bone Miner Res 2011;26(7):1381-1388.
(P761)