Diagnosing Lynch Syndrome
J Gleeson1, D Gallagher2,3
1Mater Misericordiae University Hospital, Eccles Street, Dublin
2Mater Private Hospital, Eccles Street, Dublin
3St. James’ Hospital, James’ Street, Dublin
Introduction
Lynch Syndrome, also known as Hereditary Non-Polyposis Colorectal Cancer (HNPCC), is a hereditary condition that increases an individual’s risk of developing a constellation of cancers. These most commonly arise in the colon, but also involve other solid organs such as the endometrium and ovaries in women, the stomach, brain and the skin. Ireland’s small population offers an opportunity to identify all those with Lynch Syndrome (LS) in the country, which would represent a powerful preventive opportunity to meaningfully impact on the incidence of cancer in Ireland.
There are 2,486 incident cases of colorectal cancer (CRC) in Ireland per year1, and more than one million per year worldwide. LS accounts for approximately 2-4% of these2, meaning there are up to 100 preventable cases in Ireland every year caused by LS. The average age of diagnosis of colorectal cancer in LS cases is 45 years2 (vs. 69 years in the general population). Early-onset malignancies have a particularly significant societal impact and LS-associated malignancies in younger individuals are oftentimes preventable. Efforts to improve the diagnosis of LS in Ireland requires improved physician and patient awareness of this condition and its diagnostic pathway, and a coordinated multidisciplinary national effort.
Lynch Syndrome History
LS was first described in 1913 by the American pathologist Aldred Scott Warthin.3 He published the pedigree of ‘Family G’, which included numerous cases of colorectal, uterine and gastric cancers. He became interested in the family’s history after a seamstress in the town of Ann Arbor correctly predicted her early death from cancer, even though she was healthy at the time, based on the large number of her ancestors who had died of intestinal tract and gynaecological cancers, generation after generation at a young age. Warthin traced her genealogy and searched for death reports, publishing his findings almost 20 years later. Subsequently, in 1966, Henry T. Lynch et al. reported Families ‘M’ & ‘N’ with a very similar collection of tumours to Warthin’s ‘Family G’.4 Further analyses of these three families, and other families with similar presentations, led to the recognition of Lynch Syndrome (LS).
More recent studies, facilitated by advances in molecular genetic analysis, have identified a number of genes involved in the development of LS; namely MLH1, MSH2, MSH6, and PMS2. These genes encode proteins involved in the mismatch repair (MMR) complex, which normally helps to repair DNA replication errors. A deficiency of these corrective proteins causes DNA with replication errors to proliferate (which can be detected by immunohistochemical staining). These replication errors then cause carcinogenesis in two main ways. The error can result in a gene gaining function and stimulating cell reproduction, i.e. oncogenes. Alternatively the error can cause a loss of the cells ability to stop overproduction by apoptosis, i.e. in tumour suppressor genes.
With unchecked replication errors, there is expansion or reduction in the length of repetitive DNA sequences (known as microsatellites) in the tumour DNA. These characteristic molecular changes are called microsatellite instability (MSI). MSI can be tested in most labs by performing standard polymerase chain reaction (PCR) DNA amplification and separating the amplified DNA by electrophoresis. This is performed on both tumour and normal tissue and the rates of MSI compared between the two. Tumours in LS are termed MSI high (MSI-H) due to their increased levels of microsatellite instability. Subsequently, LS is often diagnosed by identifying a germline mutation in a DNA MMR gene.
When to suspect Lynch Syndrome as a diagnosis?
CRC’s in younger patients, particularly those in the ascending colon, are suspicious for LS, as is having a synchronous tumour (either in the colon or elsewhere) at the time of diagnosis. Cancers that develop rapidly, i.e. small adenomas developing into carcinomas surprisingly quickly, would also raise suspicion for LS in the right clinical context as these tumours are known to display accelerated carcinogenesis. Patients developing multiple cancers in the typical LS sites (see below) and an autosomal dominant inheritance pattern are other indicators.
The commonest cancer seen in LS is colon cancer with predominantly mucinous or signet ring cell morphology. LS patients have a ≥70% lifetime risk of colon cancer, although this varies by genotype. Endometrial adenocarcinomas and ovarian cancers in women are typical LS-associated cancer sites. Stomach or small bowel cancers, cholangiocarcinomas and pancreatic adenocarcinomas are also typical. Urothelial transitional cell carcinomas, and CNS glioblastoma’s (Turcot’s Syndrome variant), would also be considered LS-associated cancers.
The revised Amsterdam criteria (Amsterdam II) and Bethesda criteria, then determine in whom molecular testing for LS should be considered. The Amsterdam II criteria5 require at least three relatives with a LS-associated cancer. One should be a first-degree relative to the other two, at least two successive generations should be affected and at least one should be diagnosed before age 50. The Bethesda criteria6 require diagnosis of a colorectal cancer in a patient who is less than 50 years of age or the presence of multiple primary (synchronous or metachronous) colorectal or other LS-associated tumours, regardless of age. Colorectal cancer with the MSI-H histology diagnosed in a patient who is less than 60 years of age is an alternative criterion, however including the cut-off age remains controversial.
Diagnostic Pathway
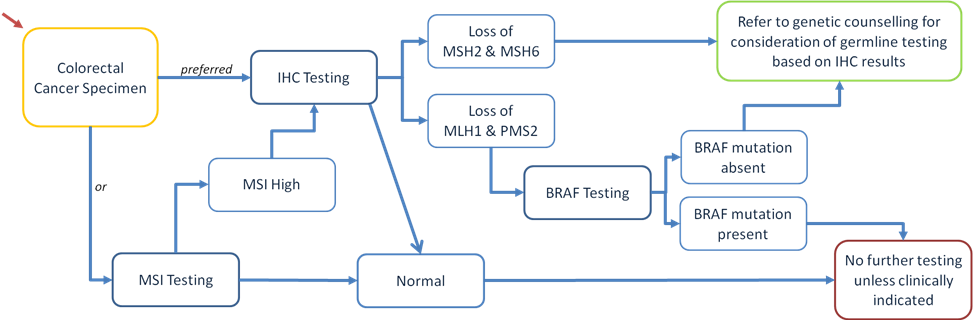
Diagram 1. Summary of the diagnostic pathway for diagnosing Lynch Syndrome. MSI – Microsatellite Instability, IHC – Immunohistochemistry.
There is a systematic approach to diagnosing LS, Diagram 1. Once a personal and family history of cancer meets the Amsterdam II and/or Bethesda criteria, the tumour should be tested for a deficiency of the MMR proteins by immunohistochemistry (IHC), or alternatively for MSI by PCR. IHC is a low-cost, informative and readily available testing method which is more specific than MSI testing. Antibodies to the MMR proteins are used as stains to detect a deficiency in the tumour cells. MSI testing is the alternative to IHC but only 20-25% of all MSI-H tumours will be positive for the germline mutations associated with LS. It is also a more expensive test and a larger quantity of tumour is needed for analysis which can be a limiting factor. Endoscopic biopsies are often insufficient. Furthermore, IHC staining has the added benefit of identifying which MMR genes should be tested for, and is therefore preferable to MSI testing initially.
Then if the MSH2 and MSH6 MMR proteins are deficient by IHC, the patient should proceed to genetic testing. These mutations are less commonly sporadic and the likelihood of a hereditary germline mutation in these genes is much higher.
MLH1 and PMS2 protein deficiencies by IHC are more likely to be due to sporadic mutations and if deficient, BRAF mutation testing is carried out next. A BRAF mutation makes LS very unlikely. Unless there is strong clinical suspicion, no further testing is required at this point if a BRAF mutation is present. However if clinically indicated, MLH1 promoter hypermethylation testing can also be carried out to identify sporadic cancers. If the BRAF mutation is absent, the likelihood of a germline defect is higher and the patient should be referred to genetic counselling for consideration of germline testing of these genes (MLH1 & PMS2).
Genetic sequencing techniques are then used to confirm the germline mutation in the DNA MMR gene identified by IHC testing. Such genetic testing in Ireland is coordinated through cancer genetics clinics in Our Lady’s Children’s Hospital Crumlin, St. James’s Hospital and the Mater Misericordiae University Hospital.
Screening for patients with Lynch Syndrome
Ireland’s small population offers an opportunity to identify all those with Lynch Syndrome (LS) in the country. This type of screening would represent a powerful preventive opportunity to meaningfully impact on the incidence of LS-associated malignancies, particularly colorectal cancer. These early onset malignancies have a significant impact in society. Screening all colorectal cancers with IHC testing, and/or MSI testing, is being introduced nationally.
Once initiated, whether due to clinical suspicion of LS or universal IHC testing, the diagnostic pathway must be pursued to completion. Ireland must develop a national infrastructure for the management of this and other molecular testing. And apart from the clinical benefit this cancer prevention example offers, there would also be considerable cost saving for the cancer care budget. The molecular testing described is relatively inexpensive, especially compared with the costs of managing cancer. This must be considered in planning for the future of Irish cancer care.
With regards individual patient surveillance, colon surveillance with full colonoscopy is vital in LS patients and has been proven to significantly reduce the cumulative risk of CRC-associated mortality. A Dutch study before and after the introduction of a screening programme for LS, demonstrated a 70% reduction in mortality with screening.7 The National Comprehensive Cancer Network (NCCN) recommends colonoscopy be carried out between the ages of 20 and 25 and then every 1-2 years in LS, or 2-5 years prior to the earliest onset of colon cancer if previously diagnosed before 25. Gastroscopy has not been shown to reduce the risk of gastro-oesophageal cancers however and only selected individuals or families should have screening every 3-5years from the age of 30.8
In women with LS, endometrium and ovary screening is also important as they have a lifetime risk of 40-60% for endometrial cancer and 12-15% for ovarian cancer. Prophylactic hysterectomy and bilateral salpingo-oophorectomy when childbearing is completed should be considered, as it significantly reduces the risk of these cancers.8 Annual screening from the age of 30-35 comprising transvaginal ultrasound (TVUS) and endometrial aspiration, as well as CA-125 is an alternative, but has not been shown to reduce mortality.
In Summary
Lynch Syndrome is underdiagnosed in Ireland. Physician and patient awareness regarding the heritability of colorectal cancer is improving but remains behind that of hereditary breast cancer. Identification of genetic predisposition to Lynch syndrome associated malignancies in a healthy population offers a powerful cancer prevention opportunity. It is important that Ireland replicates international efforts to transition healthcare to a more preventive service over the next decade. Accurate Lynch Syndrome diagnosis would provide a useful pilot for such an initiative. A nationally funded structure with specific protocols will be needed to ensure appropriate molecular testing and downstream clinical care.
Conflict of Interest:
None of the authors have any conflicts of interest to declare.
Correspondence: Dr. Jack Gleeson, Medical Oncology SpR, Beaumont Hospital, Beaumont Road, Dublin 9, Ireland.
Email: [email protected]
References
1. National Cancer Registry of Ireland, Colorectal cancer in Ireland 1994-2010. June 2013 version.
2. HT Lynch, PM Lynch, SJ Lanspa, CL Snyder, JF Lynch, CR Boland. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009 Jul; 76(1): 1–18
3. Warthin AS. Heredity with reference to carcinoma as shown by the study of the cases examined in the Pathological Laboratory of the University of Michigan, 1895-1912. Arch Int Med. 1913; 12:546–555.
4. Lynch HT, Shaw MW, Magnuson CW, Larsen AL, Krush AJ. Hereditary factors in cancer. Study of two large midwestern kindreds. Arch Intern Med. 1966;117(2):206–212.
5. Vasen H, Watson P, Mecklin JP & Lynch H. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology, 116(6), 1453–1456.
6. A Umar, CR Boland, JP Terdiman, S Syngal, A de la Chapelle, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, Lindblom A, Lynch HT, Peltomaki P, Ramsey SD, Rodriguez-Bigas MA, Vasen HF, Hawk ET, Barrett JC, Freedman AN, S Srivastava, et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J Natl Cancer Inst. 2004 Feb 18; 96(4): 261–268.
7. de Jong AE, Hendriks YM, Kleibeuker JH, de Boer SY, Cats A, Griffioen G, Nagengast FM, Nelis FG, Rookus MA, Vasen HF. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006 Mar; 130(3):665-71.
8. National Comprehensive Cancer Network guidelines: Genetic/Familial high-risk assessment: Colorectal; Lynch Syndrome guidelines. Version 2.2015
P487